An adaptive quantum chemistry workflow builds accurate molecular structures from reliable fragments, offering a practical way to study and design complex organic materials for sustainable technologies.
(Nanowerk Spotlight) Organic molecules now underlie many technologies once built around metals. They appear in organic light emitting diodes, solar cells and metal free catalysts. Their appeal comes from how small changes in structure can control color, charge flow or reaction speed. These effects depend on precise geometric details, so researchers need accurate information about bond lengths and angles in a molecule’s most stable, vibration free form.
Experimental tools such as microwave spectroscopy can measure these features by tracking how molecules rotate in the gas phase. The rotation pattern reveals atomic distances with high precision. These methods are effective for small or volatile molecules but do not extend well to larger systems, which leaves many structures without direct measurements.
Computational chemistry offers broader coverage. High accuracy quantum methods can reach the same precision as gas phase experiments, yet they slow down sharply as molecules grow. Faster methods such as many density functionals often miss subtle electron effects that influence geometry. Even small structural errors can affect predictions of light absorption or catalytic behavior.
This creates a challenge in sustainable nanomaterials, where researchers want to explore many possible structures quickly and need trustworthy models to guide that work. It also limits machine learning efforts that depend on reliable structural data.
These schemes can reproduce structural details with very small errors. The study adapts them so they can be applied to larger organic and bio inspired molecules without heavy computational cost.
Aromatic building blocks analyzed in this work: Benzoquinone (BQ), Naphtoquinone (NQ), Antraquinone (AQ), Carbazole (CBZ), Coumarin (CMR), Imidazolinone (IMD), Guaiacol (GUA), Syringol (SYR), and Styrene (STY). (Image: Reproduced from DOI:10.1002/sstr.202500557, CC BY)
Two PCS variants anchor the workflow. The first, PCS2, is a high accuracy method that describes interactions between electrons in detail and includes corrections that improve how inner and outer electrons are treated. PCS2 works well for molecules with about twenty to twenty five atoms.
The study checks PCS2 against semi experimental structures. These structures come from combining measured rotational constants with theoretical estimates of how vibrations shift those constants. PCS2 matches these benchmarks within a few thousandths of an ångström in bond lengths and about a tenth of a degree in angles.
The second variant, BDPCS3, is made for larger systems. It uses a density functional that includes an added term to capture some electron correlation, then applies small empirical corrections to certain bond types. These corrections address known limitations in the underlying method. BDPCS3 is much faster than PCS2 but needs validation to stay accurate across different chemical environments.
To handle this, the workflow analyzes molecules in fragments. It divides a structure into parts such as rings, linkers or functional groups. Each fragment is optimized with BDPCS3. If the fragment appears in a reference database, its reliability is already known. If high accuracy PCS2 data or experimental measurements exist, the workflow compares BDPCS3 results with these values after applying simple vibrational corrections.
If the difference stays within 0.1 percent, the fragment is accepted. If not, the workflow applies a correction.
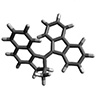
Nonaromatic building blocks analyzed in this work: Fluorene (FLU), 2-methyl-3H-cyclopenta[a]naphthalene (MCN), Molecular Motor (MOT), Pyrrolidine (PRR), Proline (PRO), Cyclohexanone Oxide (CHO). (Image: Reproduced from DOI:10.1002/sstr.202500557, CC BY)
Two correction paths exist. One uses a multilayer approach where the difficult fragment receives a high accuracy PCS2 calculation while the rest of the molecule uses the faster method. The results are combined into a single structure. The other computes the difference between PCS2 and BDPCS3 for a simplified model of the fragment and then applies that difference to the larger molecule.
The workflow proceeds through a short sequence of steps. A basic Hartree–Fock method provides a starting geometry. A hybrid functional refines vibrational properties needed for comparison with experiment. BDPCS3 supplies the main structure. PCS2 or a correction derived from it appears only for fragments that need extra accuracy. Numerical techniques help the calculations converge smoothly, which allows the entire workflow to run on standard workstation hardware.
To automate these procedures, the study introduces a tool named Nano LEGO. It identifies fragments, checks them against the reference database, requests new high accuracy data when needed and chooses the appropriate method for each part. It then rebuilds the complete molecule from validated or corrected fragments. This allows the workflow to scale to larger structures while keeping errors low.
The study tests the workflow on several molecules relevant to sustainable nanomaterials. Quinones provide rigid electron accepting structures used in organic solar cells and catalysts. A molecular motor made of a fluorene unit attached to a substituted naphthalene tests whether the workflow can handle a roughly fifty atom system whose rotation depends on precise geometry.
Heteroaromatic units such as carbazole, coumarin and imidazolinone appear in organic light emitting systems, solar cells and fluorescent proteins. Renewable aromatics such as guaiacol and syringol expand the variety of fragments in the reference set. Proline checks how the workflow handles molecules that can adopt several shapes. Styrene and a Criegee intermediate formed from cyclohexanone challenge the workflow because they include regions known to be difficult for faster quantum methods.
Across these systems, BDPCS3 with vibrational corrections predicts rotational constants within about 0.1 percent of experimental data or PCS2 references. Quinones show close agreement in bond lengths between BDPCS3 and PCS2. The molecular motor matches experimental rotational constants once vibrational effects are applied. Carbazole and coumarin align with measured values. Imidazolinone tracks its PCS2 reference accurately. Guaiacol and syringol show small deviations even though their ring substituents strongly influence electron distribution.
Proline shows that BDPCS3 can handle flexible molecules. Its simplest ring form matches PCS2, and refined energy comparisons show differences similar to small shifts arising from the motion of atoms even at their lowest energy state. This suggests that BDPCS3 captures the balance of forces that guide changes in shape.
The two most challenging systems require local corrections. In styrene, small adjustments to the bond linking the aromatic ring and the vinyl group recover agreement with PCS2 and experiment. In the Criegee intermediate, BDPCS3 underestimates the O–O bond length and distorts the C–O–O angle because these features are sensitive to electron effects that require more detailed treatment. Applying high accuracy calculations only to this portion of the molecule or using corrections derived from a simpler peroxide reduces the error by about a factor of ten.
The study shows that combining fragment level checks, selective high accuracy methods and modest corrections can produce reliable structures for a wide range of organic and bio inspired molecules. This supports the design of sustainable nanomaterials, where small geometric changes can influence how a material absorbs light, moves charge or supports chemical reactions. By balancing accuracy with efficiency, the workflow offers a practical way to explore complex molecular designs.
For authors and communications departmentsclick to open
Lay summary
Prefilled posts
ORCID information
Vincenzo Barone (National Interuniversity Consortium of Materials Science and Technology)
, 0000-0001-6420-4107 corresponding author
Nanowerk Newsletter
Get our Nanotechnology Spotlight updates to your inbox!
Thank you!
You have successfully joined our subscriber list.
Become a Spotlight guest author! Join our large and growing group of guest contributors. Have you just published a scientific paper or have other exciting developments to share with the nanotechnology community? Here is how to publish on nanowerk.com.

![Nonaromatic building blocks analyzed in this work: Fluorene (FLU), 2-methyl-3H-cyclopenta[a]naphthalene (MCN), Molecular Motor (MOT), Pyrrolidine (PRR), Proline (PRO), Cyclohexanone Oxide (CHO)](http://www.nanowerk.com/id68109_2.jpg)

